Data Set for Single Cell Transcriptomics Challenge:
Classification of COVID-19 severity using scRNA-Seq
Identification of molecular signatures of severity of COVID-19 infection has become of outmost importance for early treatment of this pandemic disease. For this the use of single-cell RNA sequencing (scRNA-Seq) makes possible to identify and quantify thousands of genes within thousands of cells. In this context, scRNA-Seq technology have called for novel artificial intelligence (AI) solutions for data analysis and medical application. The present challenge consists in the application of an AI algorithm to predict severity of COVID-19 infection using a scRNA-Seq dataset. This AI model can be of great significance and of practical value for further study of the signatures of severity of COVID-19.
Download link:
The participants will be provided with the dataset after completing the registration process and sending email to mario.flores@utsa.edu in order to obtain access.
Dataset description:
The dataset “scBALF-COVID-19” is derived from public data[1] and contains a set of Broncho Alveolar Lavage Fluid (BALF) cells from patients categorized clinically as having mild, severe or no COVID-19 infection.
- In detail, scBALF-COVID-19 contains scRNA-Seq data from three types of patients categorized clinically as having: mild infection (M# of cells), severe infection (S# of cells) or no infection (N# of cells).
- The dataset consists of one count matrix with T total number of cells and G total number of genes (Figure 1a) and a labeling matrix with labels for each cell as normal, mild or severe (Figure 1b).
- The dataset has been normalized for technical differences between patients (batch normalization) as well as sequencing depth differences between cells in each patient. This means that no normalization is required but participants may implement further normalization in order to improve results.
Dataset usage:
- We suggest utilizing the scBALF-COVID-19 dataset to develop an AI model to identify COVID-19 severity (Figure 1c).
- The dataset is imbalanced between the three classes, reflecting a practical problem in applying machine or deep learning techniques.
- The challenge encourages design of interpretable machine learning models in order to obtain putative molecular markers of COVID-19 severity.
Performance Evaluation:
- Please register your team at https://easychair.org/conferences/?conf=ibacp2021
- Each member must register at https://healthcaresummit.ieee.org/registration/
- The performance of each model will be judged by a committee of judges. The winners will be decided based on:
- Mean ROC, that is treat every label as one vs. rest and compute ROC and then average them.
- Interpretability of the developed models to identify molecular signatures.
- Results of the model classifier must be posted on: https://ieee-dataport.org/competitions/hackathon-bioinformatics-single-cell-sequencing-0
- Each competitor / group should submit through this web site an up to 4-page paper summarizing the method and results, following the standard IEEE format for conference paper submissions.
- Provide the URL to your GitHub repo, where your code can be accessed, and the prediction results can be reproduced.
- Submit the prediction results on the test set (in CSV format).
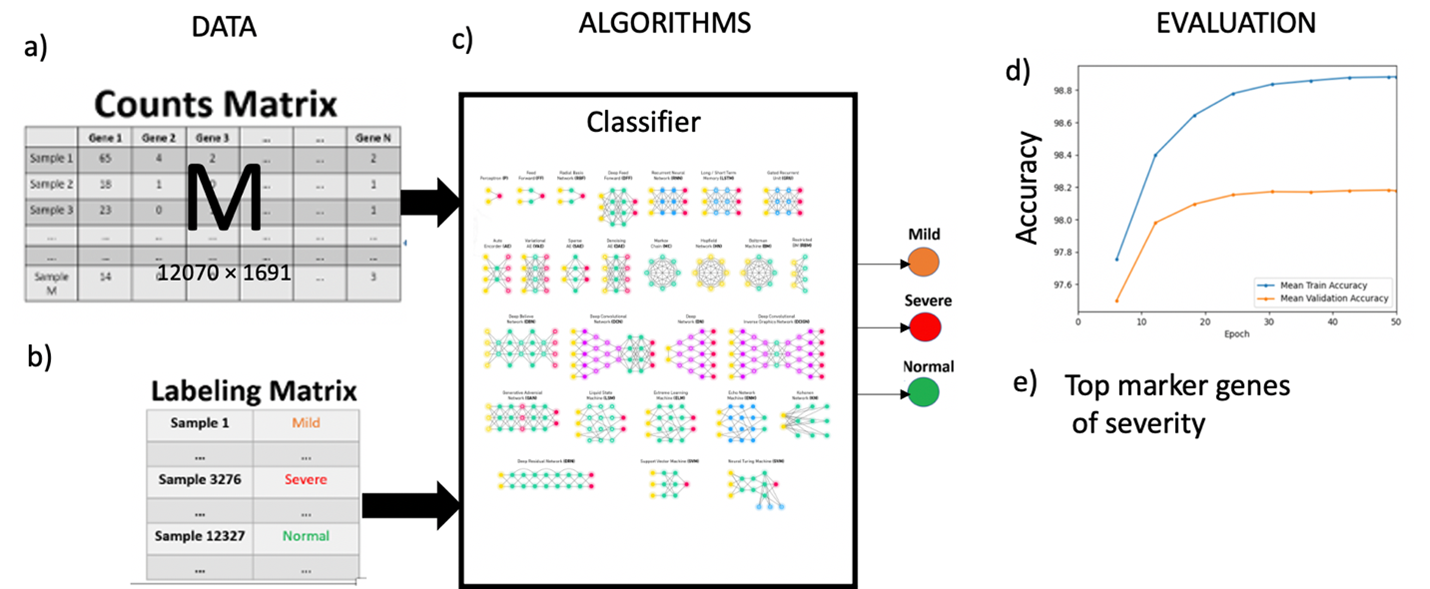
Figure 1. Classification of COVID-19 severity using scRNA-Seq.
References:
[1] M. Liao et al., “Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19,” Nat Med, vol. 26, no. 6, pp. 842-844, Jun 2020, doi: 10.1038/s41591-020-0901-9.